Overview
The AutoPLI web server is designed to fully automatically predict protein-ligand interactions.
Specifically, the user only needs to provide a query ligand structure and the UniProt ID of a target protein.
The server will collect all available protein-ligand complex structures for the target protein from current Protein Data Bank.
Then, both template-based and docking-based methods are performed to predict the query ligand-protein complex structures based on known protein-ligand complex structures.
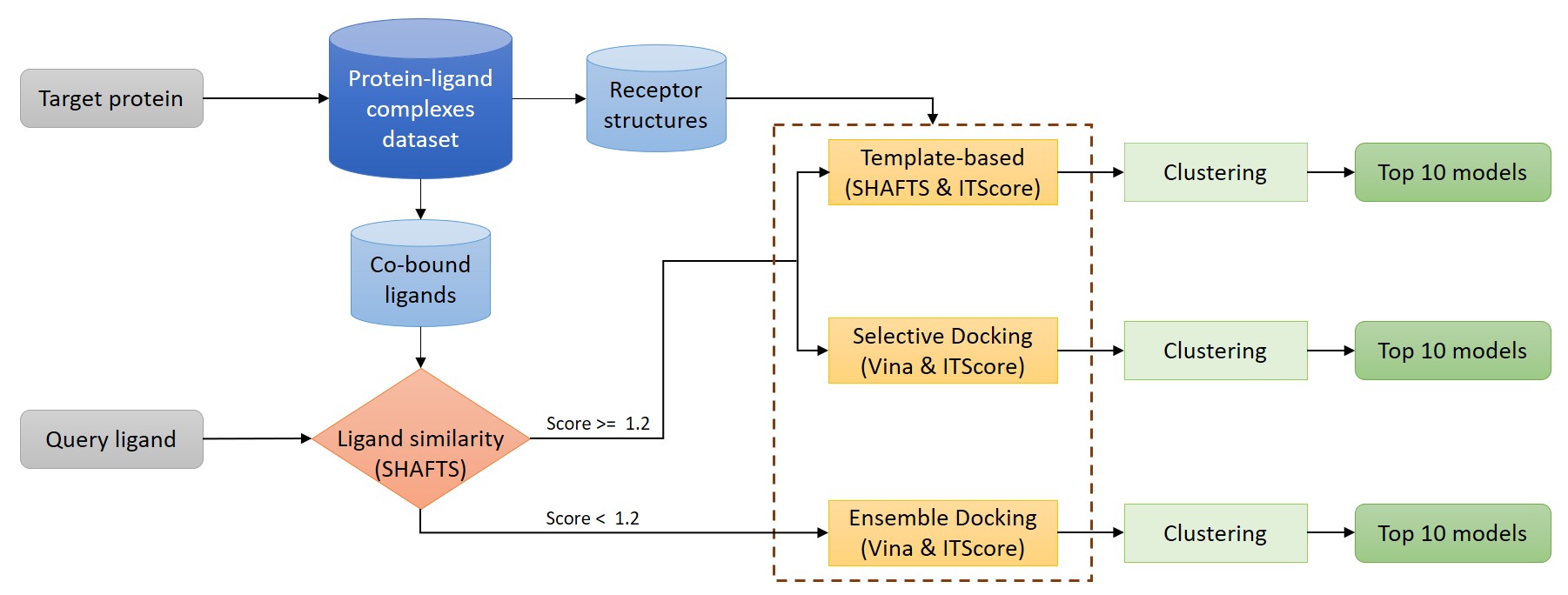
The following figure shows a flowchart of the prediction strategy used in the AutoPLI server.
Methodologies
The followng procedures will be performed by AutoPLI according to inputs from the user:
References:
Xu X, Duan R, Ma Z, Zou X. AutoPLI: A Fully Automated Server for Predicting Protein-ligand Interactions. (submitting)
Xu X, Yan C, Zou X.
Improving binding mode and binding affinity predictions of docking by ligand-based search of protein conformations: evaluation in D3R grand challenge 2015.
Journal of Computer-Aided Molecular Design, 31: 689-699, 2017.
[link]
Duan R, Xu X, Zou X.
Lessons learned from participating in D3R 2016 Grand Challenge 2: compounds targeting the farnesoid X receptor.
Journal of Computer-Aided Molecular Design, 2017 (In Press).
[link]