Introduction
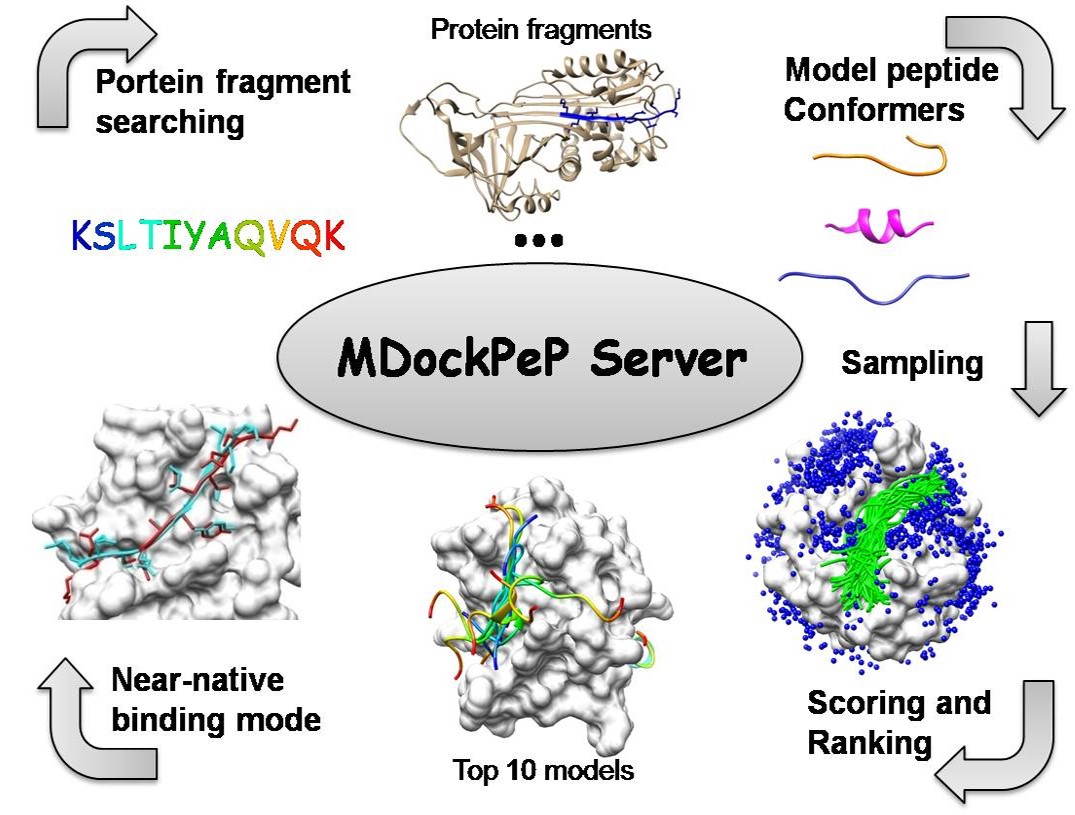 MDockPeP server predicts protein-peptide complex structures starting with the protein structure and peptide sequence.
MDockPeP server predicts protein-peptide complex structures starting with the protein structure and peptide sequence.
The prediction process consists of three steps:
(1) Modeling peptide conformers;
(2) Globally and flexibly sampling protein-peptide binding modes;
(3) Scoring and ranking the sampled binding modes.
Please go to the "About" page for the details about the MDockPeP method.
To submit a job, at least two inputs are required, the peptide sequence and the protein receptor structure.
Detailed instructions about job submition and prediction results are described on the "Tutorial" page.
Please cite the following references for MDockPeP:
- Xu X, Yan C, Zou X. MDockPeP: An ab-initio protein-peptide docking server. J Comput Chem, 39: 2409-2413, 2018. [link]
- Yan C, Xu X, Zou X. Fully Blind Docking at the Atomic Level for Protein-Peptide Complex Structure Prediction. Structure, 24: 1842-1853, 2016. [link]