About MDockPeP
Overview
The MDockPeP server predicts protein-peptide complex structures starting with the protein structure and the peptide sequence.
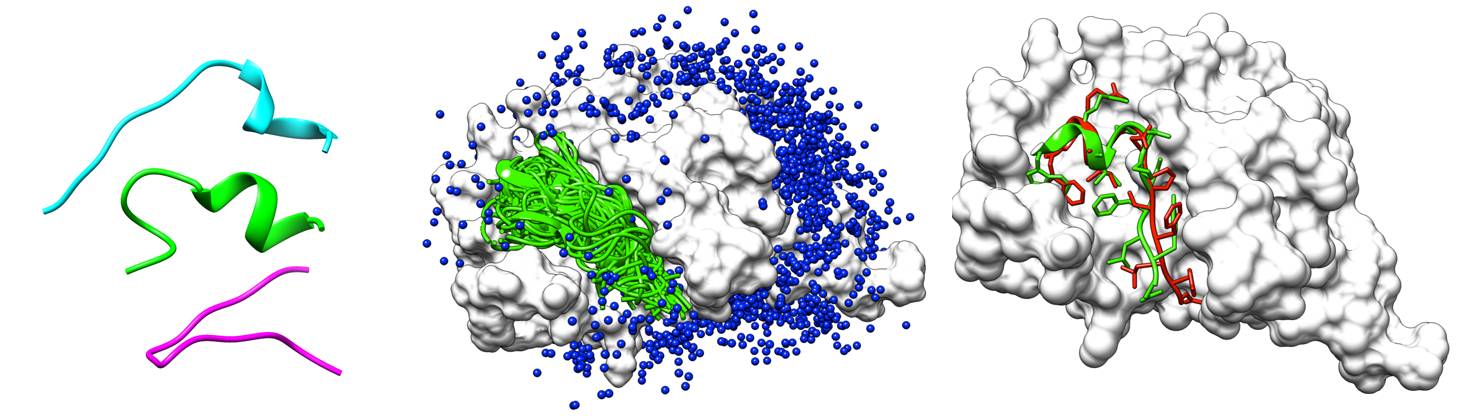
A flowchart of the protocol is shown in the figure on the right.

(1) Model peptide conformers;
(2) Sample putative peptide binding modes on the whole protein surface;
(3) Rank the sampled binding modes according to their energy scores.
Briefly, given
the sequence of the peptide and the 3D structure of the protein, first, peptide
conformers are constructed by using protein fragments with similar sequences
as the templates. According to our systematic studies, it is optimal to keep up
to (the top) three non-redundant peptide conformers for docking
calculations. The peptide conformers are then independently and flexibly
docked onto the whole protein surface using a novel iterative approach, which
samples the translational, rotational, and conformational space of the all-atom
model of each peptide conformer but keeps only those binding modes in
which their peptide conformations are relatively similar to the initial peptide
conformations (i.e., backbone RMSD or bRMSD less than 5.5 Å). The reason
behind the conformation restricition is to restrain the search space of the protein-peptide
complex configurations. Finally, the remaining binding modes are
ranked using a new statistical potential-based scoring function, ITScorePeP. ITScorePeP is
derived for protein-peptide interactions based on known protein-peptide complex
structures, using an iterative approach to circumvent the reference state
challenge.
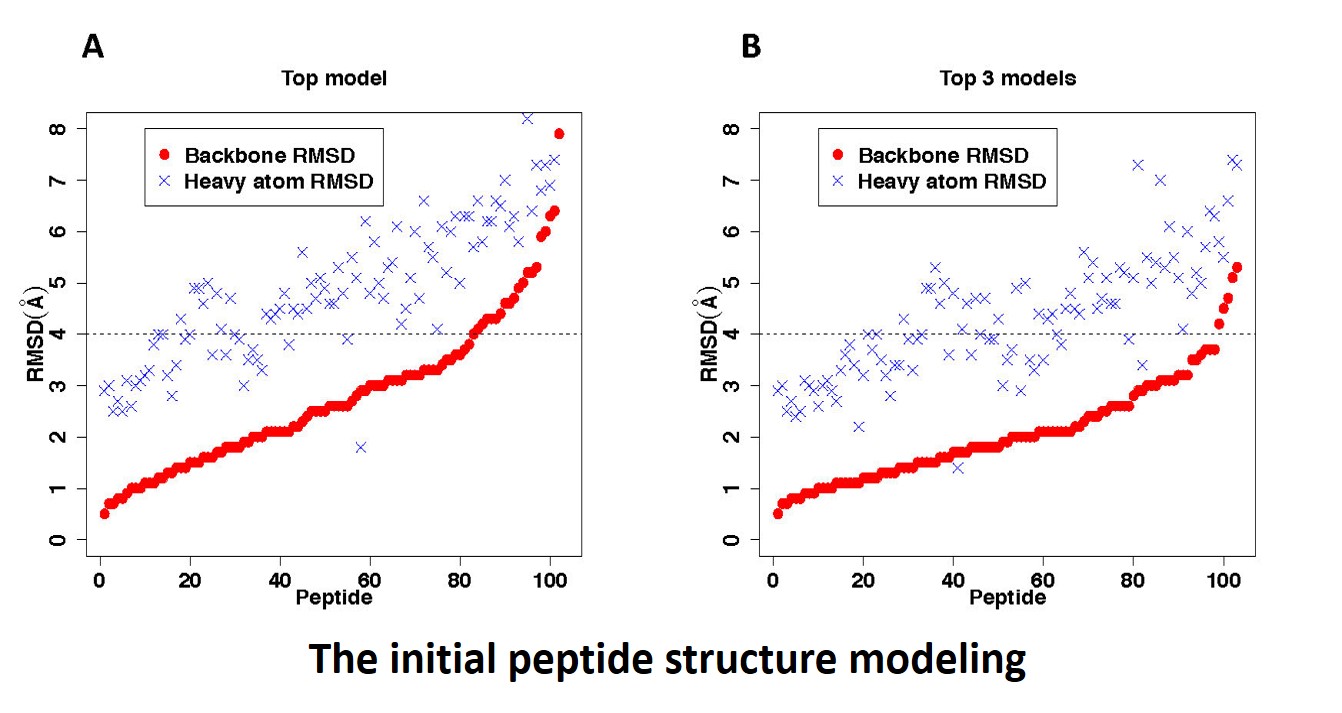
Performance on the peptiDB database
MDockPeP was validated on the peptiDB database.
In the peptide modeling step, our top conformer yields a bRMSD less than
4.0 Å for 79.6% of the cases; the median of the bRMSDs equals
to 2.6 Å. The best model of our top 3 conformers yields a bRMSD less
than 4.0 Å for 95.1% of the cases; the median value equals to 1.9
Å. The median values of the heavy-atom RMSD (hRMSD) for our top prediction and top 3 predictions are 4.8 Å and 4.3 Å, respectively.
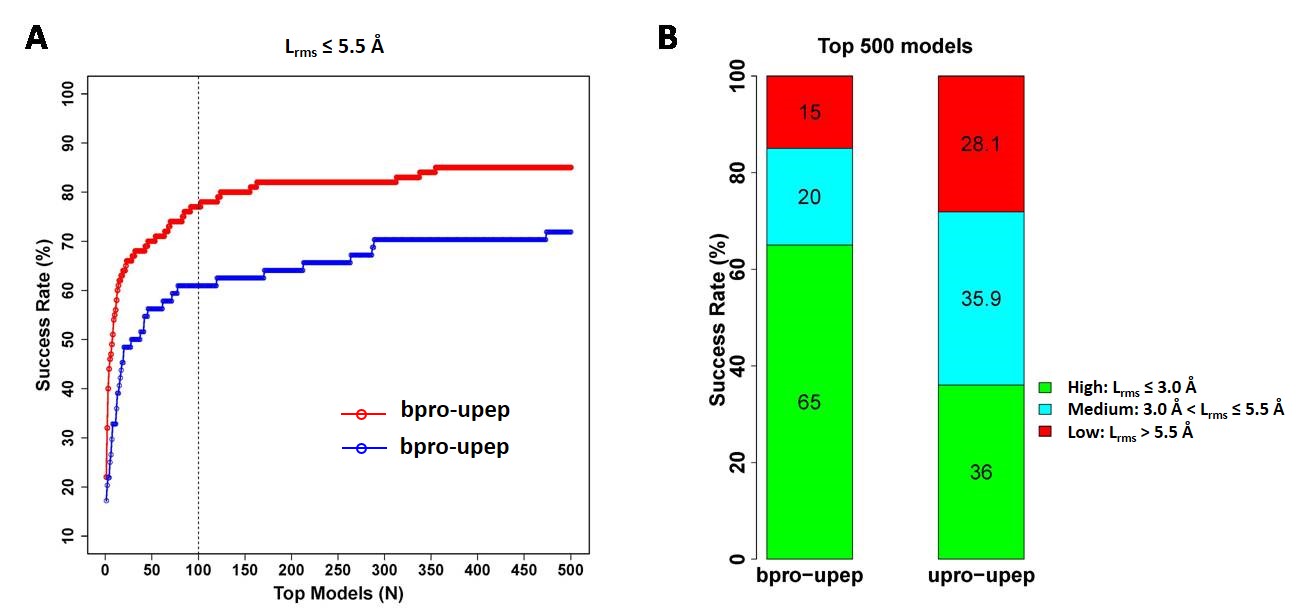
On the left panel (A) in the following figure, the success reaes of ranking at least one near native (Lrms less than 5.5 Å) mode among the top N model for the bound docking cases (bpro-upep) and the unbound docking cases (upro-upep), respectively. When considering top 500 models which are provided for the user as the sampling results, the success rate is 85% for bound docking cases (high quality: 65%; medium quality: 20%; as shown on the right panel (B)), and 71.9% for the challenging unbound docking cases (high quality: 36%; medium quality: 35.9%). The dashed balck line on the left panel corresponds to the case that N equals to 100. The corresponding success rates are 77% and 60.9% for bound and unbound dockings, respectively.
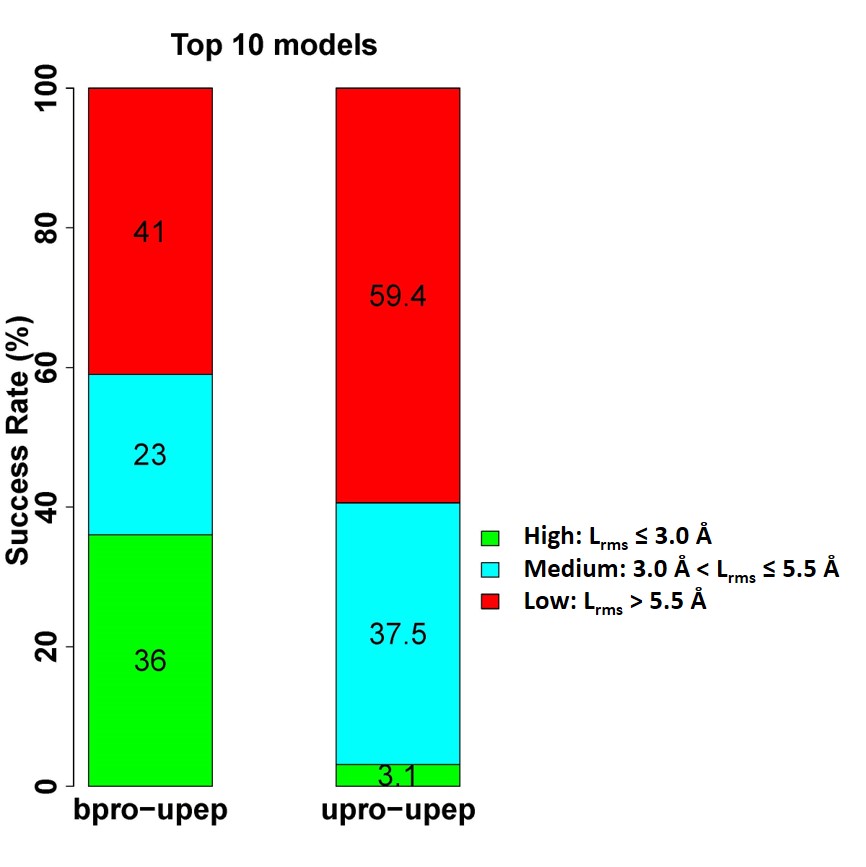
The following figure shows the performance according to top 10 modes for the bounding docking cases and unbound docking cases, respectively. The MDockPeP server successfully predicted at least one near-native mode in the top 10 models for 59% of the bound docking (high quality: 36%; medium quality: 23%), and 40.6% of the unbound docking (high quality: 3.1%; medium quality: 37.5%).
MDockPeP is computationally efficient, and normally takes 1 to 10 hours on a node of 24 Intel Xeon cores (CPU E5-2650 v3 @ 2.30GHz) for each docking depending on the sizes of the peptide and the protein.
More details about MDockPeP are available in the following references: